Phasing을 위해서 Li and Stephens model (LSM)을 사용한다. LSM의 계산속도를 높이기 위해서, 주어진 genotype에 대해서 가능한 haplotype을 만들어 Positional Burrows-Wheeler Transform (PBWT) 과정을 진행한다. PBWT에 대해 잘 정리된 글이 있어서 이해한 내용을 정리해 본다.
# Haplotypes
X = [[0, 1, 0, 1, 0, 1], # 0
[1, 1, 0, 0, 0, 1], # 1
[1, 1, 1, 1, 1, 1], # 2
[0, 1, 1, 1, 1, 0], # 3
[0, 0, 0, 0, 0, 0], # 4
[1, 0, 0, 0, 1, 0], # 5
[1, 1, 0, 0, 0, 1], # 6
[0, 1, 0, 1, 1, 0]] # 7
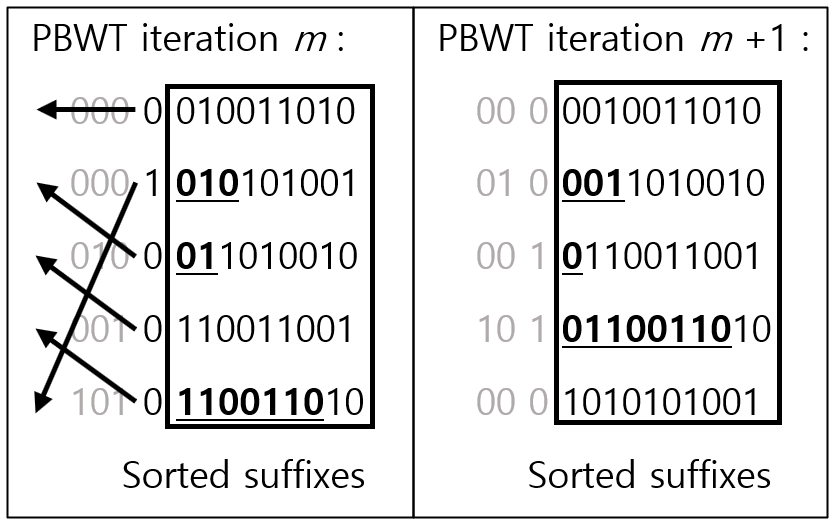
기본적인 아이디어는 haplotype의 reversed prefix의 순서대로 정렬하여 match되는 가장 긴 genotype을 찾는 방법이다.
haplotype의 맨 끝 genotype을 제외하고 0의 갯수가 많은 것부터 정렬한다. 0의 갯수가 동일하면 0이 뒤쪽에서 먼저 오는 것이 선순위가 된다. 정렬된 haplotype 순서에서 바로 앞의 haplotype과 match되는 genotype을 찾아 가장 긴 match를 찾을 수 있다. 정렬하여 match된 genotype은 밑줄로 표시하였다. 이렇게 reversed order로 정렬을 하게 되면, 가장 길게 match되는 sequence는 가까이(adjacent) 오게 되어있다.
> display_prefix_and_divergence_arrays(X)
4 | 00000 0
1 | 11000 1
6 | 11000 1
0 | 01010 1
5 | 10001 0
7 | 01011 0
3 | 01111 0
2 | 11111 1
다음은 haplotype이 주어지면, 최소한의 haplotype으로 가장 길게 match되는 genotype sequence를 찾아주는 것이다.
> display_set_maximal_matches(X)
set maximal matches for haplotype 0:
0|010101
7|010110
set maximal matches for haplotype 2:
2|111111
1|110001
6|110001
3|011110
set maximal matches for haplotype 3:
3|011110
0|010101
7|010110
2|111111
set maximal matches for haplotype 4:
4|000000
0|010101
3|011110
7|010110
5|100010
1|110001
6|110001
set maximal matches for haplotype 5:
5|100010
1|110001
2|111111
6|110001
4|000000
set maximal matches for haplotype 7:
7|010110
0|010101
Example : Set maximal matches
> import random
> M = 3
> N = 30
> Y = [[random.randint(0, 1) for _ in range(N)] for _ in range(M)]
> display_set_maximal_matches(Y)
set maximal matches for haplotype 0:
0|100001010111100110111010100101
2|011000110001000111111011110011
1|011011110111001111100110000010
1|011011110111001111100110000010
2|011000110001000111111011110011
2|011000110001000111111011110011
1|011011110111001111100110000010
2|011000110001000111111011110011
1|011011110111001111100110000010
set maximal matches for haplotype 1:
1|011011110111001111100110000010
2|011000110001000111111011110011
0|100001010111100110111010100101
2|011000110001000111111011110011
0|100001010111100110111010100101
2|011000110001000111111011110011
2|011000110001000111111011110011
0|100001010111100110111010100101
0|100001010111100110111010100101
2|011000110001000111111011110011
set maximal matches for haplotype 2:
2|011000110001000111111011110011
1|011011110111001111100110000010
0|100001010111100110111010100101
1|011011110111001111100110000010
1|011011110111001111100110000010
0|100001010111100110111010100101
1|011011110111001111100110000010
0|100001010111100110111010100101
0|100001010111100110111010100101
1|011011110111001111100110000010
CODE : Build prefix and divergence arrays
def build_prefix_array(X):
# M haplotypes
M = len(X)
# N variable sites
N = len(X[0])
# initialise positional prefix array
ppa = list(range(M))
# iterate over variants
for k in range(N-1):
# setup intermediates
a = list()
b = list()
# iterate over haplotypes in reverse prefix sorted order
for index in ppa:
# current haplotype
haplotype = X[index]
# allele for current haplotype
allele = haplotype[k]
# update intermediates
if allele == 0:
a.append(index)
else:
b.append(index)
# construct the new positional prefix array for k+1 by concatenating lists a and b
ppa = a + b
return ppa
def display_prefix_array(X):
from IPython.display import display_html
ppa = build_prefix_array(X)
html = '<pre style="line-height: 100%">'
for index in ppa:
haplotype = X[index]
html += str(index) + '|' + ''.join(str(allele) for allele in haplotype[:-1])
html += ' ' + str(haplotype[-1]) + '<br/>'
html += '</pre>'
display_html(html, raw=True)
def build_prefix_and_divergence_arrays(X):
# M haplotypes
M = len(X)
# N variable sites
N = len(X[0])
# initialise positional prefix array
ppa = list(range(M))
# initialise divergence array
div = [0] * M
# iterate over variants
for k in range(N-1):
# setup intermediates
a = list()
b = list()
d = list()
e = list()
p = q = k + 1
# iterate over haplotypes in reverse prefix sorted order
for index, match_start in zip(ppa, div):
# current haplotype
haplotype = X[index]
# allele for current haplotype
allele = haplotype[k]
# update intermediates
if match_start > p:
p = match_start
if match_start > q:
q = match_start
# update intermediates
if allele == 0:
a.append(index)
d.append(p)
p = 0
else:
b.append(index)
e.append(q)
q = 0
# construct the new arrays for k+1 by concatenating intermediates
ppa = a + b
div = d + e
return ppa, div
def display_prefix_and_divergence_arrays(X):
from IPython.display import display_html
ppa, div = build_prefix_and_divergence_arrays(X)
html = '<pre style="line-height: 100%">'
for index, match_start in zip(ppa, div):
haplotype = X[index]
html += str(index) + '|'
for k, allele in enumerate(haplotype[:-1]):
if match_start == k:
html += '<strong><u>'
html += str(allele)
if match_start < len(haplotype) - 1:
html += '</u></strong>'
html += ' ' + str(haplotype[-1]) + '<br/>'
html += '</pre>'
display_html(html, raw=True)
def main():
X = [[0, 1, 0, 1, 0, 1],
[1, 1, 0, 0, 0, 1],
[1, 1, 1, 1, 1, 1],
[0, 1, 1, 1, 1, 0],
[0, 0, 0, 0, 0, 0],
[1, 0, 0, 0, 1, 0],
[1, 1, 0, 0, 0, 1],
[0, 1, 0, 1, 1, 0]]
ppa, div = build_prefix_and_divergence_arrays(X)
print(ppa, div)
if __name__=='__main__':
main()
CODE : Set maximal matches
def report_set_maximal_matches(X):
# M haplotypes
M = len(X)
# N variable sites
N = len(X[0])
# initialise positional prefix array
ppa = list(range(M))
# initialise divergence array
div = [0] * M
# iterate over variants
for k in range(N):
# sentinel
div.append(k+1)
for i in range(M):
m = i - 1
n = i + 1
match_continues = False
if div[i] <= div[i+1]:
# match to previous neighbour is longer, scan "down" the haplotypes (decreasing indices)
while div[m+1] <= div[i]:
if X[ppa[m]][k] == X[ppa[i]][k]:
match_continues = True
break
m -= 1
if match_continues:
continue
if div[i] >= div[i+1]:
# match to next neighbour is longer, scan "up" the haplotypes (increasing indices)
while div[n] <= div[i+1]:
if X[ppa[n]][k] == X[ppa[i]][k]:
match_continues = True
break
n += 1
if match_continues:
continue
for j in range(m+1, i):
if div[i] < k: # N.B., avoid 0 length matches, not in Durbin (2014)
yield ppa[i], ppa[j], div[i], k
for j in range(i+1, n):
if div[i+1] < k: # N.B., avoid 0 length matches, not in Durbin (2014)
yield ppa[i], ppa[j], div[i+1], k
# build next prefix and divergence arrays
if k < N - 1:
# setup intermediates
a = list()
b = list()
d = list()
e = list()
p = q = k + 1
# iterate over haplotypes in prefix sorted order
for index, match_start in zip(ppa, div):
# current haplotype
haplotype = X[index]
# allele for current haplotype
allele = haplotype[k]
# update intermediates
if match_start > p:
p = match_start
if match_start > q:
q = match_start
# update intermediates
if allele == 0:
a.append(index)
d.append(p)
p = 0
else:
b.append(index)
e.append(q)
q = 0
# construct the new arrays for k+1
ppa = a + b
div = d + e
def display_set_maximal_matches(X):
from IPython.display import display_html
from operator import itemgetter
from itertools import groupby
html = '<pre style="line-height: 100%">'
matches = sorted(report_set_maximal_matches(X), key=itemgetter(0, 2))
for i, sub_matches in groupby(matches, key=itemgetter(0)):
html += 'set maximal matches for haplotype %s:<br/><br/>' % i
hi = X[i]
html += str(i) + '|' + ''.join(map(str, hi)) + '<br/>'
for _, j, k1, k2 in sub_matches:
hj = X[j]
html += str(j) + '|' + ''.join(map(str, hj[:k1]))
html += '<strong><u>' + ''.join(map(str, hj[k1:k2])) + '</u></strong>'
html += ''.join(map(str, hj[k2:])) + '<br/>'
html += '<br/>'
html += '</pre>'
display_html(html, raw=True)
CODE : Set maximal matches from a new sequence z to X
%load_ext Cython
%%cython
import numpy as np
cimport numpy as np
def build_pbwt(np.int8_t[:, :] H):
cdef:
Py_ssize_t N, M, k, i, u, v, p, q
np.uint32_t index, match_start
np.int8_t allele
np.int8_t[:, :] pbwt
np.uint32_t[:, :] ppa, div
np.uint32_t[:] a, b, d, e
# expect haplotype data transposed
N, M = H.shape[:2]
# setup pbwt
pbwt = np.empty_like(H)
# setup positional prefix array
ppa = np.empty((N, M), dtype='u4')
# initialise first ppa column
for i in range(M):
ppa[0, i] = i
# setup divergence array
div = np.zeros((N, M), dtype='u4')
# setup intermediates
a = np.zeros(M, dtype='u4')
b = np.zeros(M, dtype='u4')
d = np.zeros(M, dtype='u4')
e = np.zeros(M, dtype='u4')
# iterate over variants
for k in range(N):
# setup intermediates
u = v = 0
p = q = k + 1
# iterate over haplotypes in reverse prefix sorted order
for i in range(M):
# index for current haplotype
index = ppa[k, i]
# match start position for current haplotype
match_start = div[k, i]
# allele for current haplotype
allele = H[k, index]
# update output
pbwt[k, i] = allele
# update intermediates
if match_start > p:
p = match_start
if match_start > q:
q = match_start
# update intermediates
if allele == 0:
a[u] = index
d[u] = p
u += 1
p = 0
else:
b[v] = index
e[v] = q
v += 1
q = 0
if k < N - 1:
# construct the new positional prefix array for k+1
ppa[k+1, :u] = a[:u]
ppa[k+1, u:] = b[:v]
# construct the new divergence array for k+1
div[k+1, :u] = d[:u]
div[k+1, u:] = e[:v]
return np.asarray(pbwt), np.asarray(ppa), np.asarray(div)
Reference
- http://alimanfoo.github.io/2016/06/21/understanding-pbwt.html
- Efficient haplotype matching and storage using the positional Burrows–Wheeler transform (PBWT), https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3998136/
- Accurate, scalable and integrative haplotype estimation, https://www.nature.com/articles/s41467-019-13225-y
'Study' 카테고리의 다른 글
| Phasing (0) | 2020.06.17 |
|---|---|
| Random Walk (0) | 2020.06.17 |
| Monte Carlo simulation (0) | 2020.06.17 |
| Hilbert curve (0) | 2020.06.01 |
| CNN terminology (0) | 2020.05.27 |



댓글